Pjotr's rotating BLOG (older stuff)
Table of Contents
- 1. In Madison
- 2. Software deployment
- 3. cupylmm (part 3)
- 4. cupylmm (part 2)
- 5. cupylmm
- 6. scikit.cuda.linalg
- 7. OmicABEL
- 8. CUDA matrix multiplication on the Tesla K20
- 9. CUDA tests
- 10. Arvados GATK is fast
- 11. First days in Memphis TN
- 12. Installing CUDA on Penguin
- 13. Arvados
- 14. The webserver environment
- 15. Fixing the error
- 16. Replicating the error
- 17. Fixing bugs
- 18. Changing environments
- 19. Running pylmm/GN2 on Penguin
- 20. Installing Guix on GN2/Penguin
- 21. Avoiding 3x extraneous genotype matrix transpose
- 22. Optimizing pylmm/GN2
- 23. pylmm home stretch (0.50 release)
- 24. Genotype normalization problem
- 25. Starting up again (Sambamba on Google OSS BLOG)
- 26. RSS feed fix
- 27. Plot of timings
- 28. Preparing for adding Human GWAS support
- 29. Adding the progress meter
- 30. Adding multi-core GWAS into GN2
- 31. Performance stuff and adding multi-core Kinship into GN2
- 32. GN2/pylmm kinship calculation
- 33. More FaST-LMM vs pylmm
- 34. GN2/pylmm pull request
- 35. pylmm vs FaST-LMM
- 36. PLINK file formats
- 37. convertlmm.py
- 38. CUDA, TESLA K20, K40 and K80 and Fixing the RSS feed
- 39. Test framework GN2/pylmm and runlmm.py entry point
- 40. Moving again on GN2/pylmm
- 41. A virtual group
- 42. Some work at KEMRI
- 43. Setting up a test framework for pylmm in GN2
- 44. Parallel programming is hard
- 45. Merging pylmm updates into GN2
- 46. Passing parameters with Redis
- 47. Google Summer of Code (GSoC)
- 48. Figuring out the logic of pylmm in genenetwork
- 49. VPN and access to a CUDA machine
- 50. More GPU and also Pacbio long reads for SV
- 51. pyCUDA
- 52. FaST-LMM the basics
- 53. Pypy and pylmm first
- 54. Pypy thoughts and running the profiler
- 55. Final speedups for pylmmGWAS and pylmmKinship!
- 56. GWAS and more on kinship
- 57. Looking at missing data and the Kinship calculation got faster
- 58. Added parallel processing for the GWAS script
- 59. First Python code for parallel processing
- 60. Working on Sambamba paper
- 61. Running pylmm
- 62. Reading on LMM (2)
- 63. Reading on LMM (1)
- 64. Started on FaST-LMM
- 65. A rotating BLOG to keep track of things
Tis document describes Pjotr's journey in introducing a speedy LMM resolver for GWAS for GeneNetwork.org.
1 In Madison
News flash GNU Guix has just gotten BLAS support for scipy. And Guix Ruby got bundler support! And we created a mailing list for bio-packaging.
Alexey started porting R/lme4 to Ruby. I hope this becomes a reference implementation of lme4 as the R code is too hard to read.
David Thompson added bundler support for Ruby in GNU Guix. I am testing it right now. I also have found a reproducible way of getting Guix running from git source, that should speed development up.
I ran bundler with success using the latest. It requires a tweak to the environment so the GEMPATH points at
$HOME/.guix-profile/lib/ruby/gems/2.2.0/
The script I use to run Ruby is here.
that essentially replaces the old rvm or rbenv! So much better :). More documentation at guix-notes.
2 Software deployment
I am on the road (Pittsburgh heading for Chicago right now) which means I work on software deployment (that is something I can always do with half a brain). First I started playing with Docker, but Docker (again) fails to charm me. It really is too much of a hack and not a true solution for software deployment. Next I deployed the GNU Guix build system on Penguin adding instructions to [guix-notes]. For the first time I got it to build with GNU Guix tools on Debian. The initial target is to build the ldc D compiler through GNU Guix - as I am bound to start using that once that works. Then for practise I'll add a sambamba/biod package. Later move on to the GN2 software stack, including CUDA.
3 cupylmm (part 3)
After some thinking I realised the parallel CUDA version is probably not working because the multi-core version of pylmm uses different processes. This may mean that garbage collection is not working and inter-process sharing may have issues too. I should look into this if we want to make it work (but it is a lesser priority).
CUDA is now used by default for the Kinship calculation. It should fall back on non-CUDA if there is a problem (no CUDA support or out of GPU RAM). I added three tweakable parameters, for the matrix sizes that will (1) switch Kinship from CPU to CUDA, (2) switch from single matrix CPU to iterable multi-core CPU and (3) switch GWAS from multi-core CPU to single-core CUDA.
The regression tests work for this setup on a machine with CUDA and without CUDA hardware. The old style GN2 support is now obsolete - handling of missing data and normalization should be handled by pylmm internally. The next step is to
3.1 IN PROGRESS integrate pylmm CUDA with GN2 using runlmm
I am in Memphis for 4 more days. Will divide my time between Arvados, pylmm, a talk I am giving and some programming discussions (today Python closures).
Note that for many items below the status has been switched to green (either DONE or DEFERRED).
4 cupylmm (part 2)
Got CUDA to work with pylmm in the single threaded version.
Under the hood Python numpy matrices can be oriented in two ways (by column or by vector) depending on the last action/transformation. A stride is a tuple where the first number represents the number of bytes you have to move to get to the next row. The second number represents the number of bytes you have to move to get to the next column. This is to optimize processing speed. A matrix transformation simple reverses the strides. When the first stride is larger than the second we have a matrix in C orientation. Otherwise the matrix is in FORTRAN orientation. This orientation is stored as a flag named ccontiguous and fcontiguous respectively. Now (apparently) both can also represent a strided array with gaps, i.e., the stride is possibly larger than the actual data represented.
The problem was that when the strides are equal the logic failed in scikits.cuda. When the strides are equal it is always a matrix with column width 1 (i.e., a vector in matrix form). I worked around it by using num.dot for these specific cases (no reason to send that to GPU anyway). I may do the same for small matrices anyway. But that requires more tuning.
The multi-threaded version is not working with CUDA. Not yet sure what is happening - I even tried a locking mechanism, but the multi-core CUDA version sits and waits forever.
Anyway the CUDA version calculates a kinship matrix in 1 second for 8000 snps x 1217 inds. The fastest full GWAS calculation is now down to 7 seconds (that what used to take 48 seconds). Even in single threaded mode the CUDA version is expected to be much faster for larger matrices than the multi-core version, so I am going to leave it like this for the time being. Now it needs further integration with GN2 and I should start work more on pipelines. Further tweaking may come between jobs.
4.1 DONE Automate CUDA -> numpy fallback when GPU runs out of RAM
5 cupylmm
Still having trouble with matrix multiplication. Let's go over this slowly. Looking at the relevant section of scikits:
a_shape = a_gpu.shape b_shape = b_gpu.shape cublas_func = cublas.cublasDgemm alpha = np.float64(alpha) beta = np.float64(beta)
it get interesting with order:
a_f_order = a_gpu.strides[1] > a_gpu.strides[0] b_f_order = b_gpu.strides[1] > b_gpu.strides[0]
Now the problem is that
a (1, 1217) [ [-0.3 -0.466 -1.237 ..., -0. 0. -0. ] ] b (1217, 1) = [ [-0.3] ... [-0.3] [-0.466] ... [-0.466] ... [ 0.] ... [ 0.] [-0.] ... [-0.] ] a_gpu strides (8, 8) b_gpu strides (8, 8) a_f_order False b_f_order False
returns an error 'On entry to DGEMM parameter number 10 had an illegal value' which is due to the fact that the number of strides for a is actually wrong. Correct working versions look like:
a (1, 1217) [ [-0. -0. -0.001 ..., -0. 0. -0. ] ] b (1217, 1) = [ [ 0.062] ... [ 0.062] [-1.199] ... [-1.199] ... [ 0.703] ... [ 0.703] [-0.427] ... [-0.427] ] a_gpu strides (9736, 8) b_gpu strides (8, 8) a_f_order False b_f_order False a (1217, 1) = [ [-0.3] ... [-0.3] [-0.466] ... [-0.466] ... [ 0.] ... [ 0.] [-0.] ... [-0.] ] b (1, 1) [ [ 0.023] ] a_gpu strides (8, 8) b_gpu strides (8, 8) a_f_order False b_f_order False
Strides is wrong probably because of a reshape command somewhere in pylmm. I found one culprit to be a numpy transpose, reflecting this. Basically, the data is stored in row-major order, i.e., consecutive elements of the rows of the array are contiguous in memory. When doing a transpose only the strides are changed. But there is more to it because the stride checking in scikits/linalg.py#L464 and L466 are plain wrong when one matrix has equal strides. Disabling these checks makes the code work. I raised an issue for that with scikits.
All this pain is due to (1) numpy having multiple data representations in memory and (2) dgemm and the CUDA version only allow data to be passed in order. Which happend to differ between implementation. It is kinda logical because the low level functions want data presented in an optimal and certain way. Anyway, it looks like it mostly runs now.
Meanwhile Alexej found lme4 in pure R which takes account of phylogeny. Interesting and (for once) readable R code.
6 scikit.cuda.linalg
I struggled a bit with the scikit.cuda libraries. They are nice because they take the pain out of moving data from RAM to GPU space (and back) and cleanly call into pycuda. Unfortunately the stable release of that library had a bug - and only after installing the latest version from git I discovered after quite a few hours that the matrix orientation differs from that used by numpy.dot (grrr). So, actually it may be a feature. Reading the CUDA docs it turns out that the matrices should be oriented that the columns of A should equal rows of B.
Anyway, the CUDA matrix dot product appears to work now as expected and I can start tuning pylmm on Penguin's K20. There is no doubt that the GPU will outperform the CPU version. A Tesla K80 will make it even better.
Btw the issue trackers on github are now being used! We are heading for a GO LIVE of Genenetwork2 soon.
7 OmicABEL
Today I moved pylmm\gn2 back into its own github repository.
Following Artem's research I took a look at OmicABEL which appears to outperform FaST-LMM especially for multiple phenotypes. This looks like something we should use in GN2. There is a GPU version too. It may well replace pylmm or be an alternative to that implementation. It is also interesting that FaST-LMM may be not much of an alternative to pylmm. This means at this stage, in addition to improving pylmm, we should:
7.1 TODO Get OmicABEL to compile and run
On Sandy Bridge the multi-core GotoBLAS2 only builds with the unoptimized
make TARGET=FORCE_GENERIC
[[https://github.com/xianyi/OpenBLAS][OpenBLAS] is the new version of GotoBLAS2 and supports the Sandy Bridge. Now, since OmicABEL was last updated in 2013 it does not compile with OpenBLAS. So that job goes on the TODO list.
7.2 TODO Get the GPU version of OmicABEL to compile and run
8 CUDA matrix multiplication on the Tesla K20
scikits.cuda does matrix multiplication on the Tesla K20. It required installing
pip install scipy pip install scikits.cuda
The RAM requirement is calculated by taking the sizes of the matrix and its transpose plus room for the resulting matrix timex the floating point size. The largest I could fit was
X=4000,Y=150000 float size=4 (4.864000 GB)
Results are encouraging:
Pump to GPU
('Time elapsed ', 2.2444241046905518)
Calculate
[[ 37488.19921875 37473.6171875 37435.11328125 ..., 37496.45703125
37452.92578125 37401.33203125]
[ 37497.21875 37425.7578125 37493.21875 ..., 37514.40625
37549.1640625 37447.3359375 ]
[ 37579.62109375 37534.2265625 37548.5 ..., 37556.26953125
37567.5625 37530.77734375]
...,
[ 37585.26171875 37544.296875 37589.78515625 ..., 37605.17578125
37592.16015625 37493.515625 ]
[ 37503.35546875 37549.12109375 37499.13671875 ..., 37531.83984375
37544.125 37583.734375 ]
[ 37406.6640625 37442.05078125 37435.39453125 ..., 37433.3359375
37418.74609375 37354.92578125]]
(4000, 4000)
('Time elapsed ', 4.7880260944366455)
which is about 100x faster than the numpy single CPU version. The code looks like:
import pycuda.gpuarray as gpuarray
import pycuda.autoinit
import numpy as np
import scikits.cuda.linalg as linalg
np.random.seed(1)
x=4000
y=150000
fpsize=4
print("X=%d,Y=%d float size=%d (%f GB)" % (x,y,fpsize,(2.0*float(x)*float(y)+float(x)*float(x))*fpsize/1000000000.0))
linalg.init()
a = np.asarray(np.random.rand(x,y), np.float32)
b = np.asarray(np.random.rand(y,x), np.float32)
a_gpu = gpuarray.to_gpu(a)
b_gpu = gpuarray.to_gpu(b)
d_gpu = linalg.dot(a_gpu, b_gpu)
Pretty straightforward stuff.
9 CUDA tests
All the NVIDA CUDA tests I try work on Penguin. That is good, now I can (carefully) test the Python toolchain again.
penguin:~/NVIDIA_CUDA-7.0_Samples/0_Simple/matrixMulCUBLAS$ export PATH=/usr/local/cuda/bin:$PATH penguin:~/NVIDIA_CUDA-7.0_Samples/0_Simple/matrixMulCUBLAS$ export LD_LIBRARY_PATH=/usr/local/cuda/lib64:$LD_LIBRARY_PATH penguin:~/NVIDIA_CUDA-7.0_Samples/0_Simple/matrixMulCUBLAS$ ./matrixMulCUBLAS [Matrix Multiply CUBLAS] - Starting... GPU Device 0: "Tesla K20Xm" with compute capability 3.5 MatrixA(320,640), MatrixB(320,640), MatrixC(320,640) Computing result using CUBLAS...done. Performance= 1215.18 GFlop/s, Time= 0.108 msec, Size= 131072000 Ops Computing result using host CPU...done. Comparing CUBLAS Matrix Multiply with CPU results: PASS
I rebuilt pycuda and friends (this time without the Guix version of Python in the mix) and this time it works fine!
. ~/guix_python/bin/activate . export CUDA_ROOT=/usr/local/cuda-7.0 export PATH=$CUDA_ROOT/bin:$PATH export LD_LIBRARY_PATH=$CUDA_ROOT/lib64 python dump_properties.py 1 device(s) found. Device #0: Tesla K20Xm Compute Capability: 3.5 Total Memory: 5897792 KB (~5GB) ASYNC_ENGINE_COUNT: 2
10 Arvados GATK is fast
We have run the basic BWA+GATK pipeline on Arvados. It took some time to get GATK/Queue ported, mostly because we needed an adaptor for submitting jobs and Queue wants a shared drive to collate results of the Queue runners and that concept is not really a good model in the Cloud (in truth, it is rather a bad idea to use a shared NFS mount anyway).
I think the results speak for themselves. For a 70GB fastq file the total processing time was 1d and 10hrs (without Queue it was 3d and 11hrs).
- bwa runs in 11h35m resulting in a 322GB SAM file
- sambamba sam2bam runs in 3h49m resulting in a 92GB BAM file
- sambamba sort runs in 3h55min
- sambamba index runs in 1h0min
- GATK haplotype caller runs in 1h59m resulting in 982Mb VCF
- GATK annotate runs in 11h47min resulting in 1.0GB VCF
Speeds can be improved further by combining sambamba functions (sort can produce an index) and sam2bam should pipe into sort. bwa can be chunked too, at least at the chromosome level. I think we can easily run this pipeline within 24hrs.
Between our run and that of Arvados the variant calling and annotation are practically identical (I checked by eye picking random variants). The number of variants on your calls count:
hpcs01:/hpc/cog_bioinf/data/mapping/external/STE105blood$ wc -l STE105blood.raw_variants.vcf 4928661 STE105blood.raw_variants.vcf
And the number from Arvados almost agree:
selinunte:~/Downloads$ wc -l STE0105blood_H0D8N_L008_R1.fastq.sorted.* 4939885 STE0105blood_H0D8N_L008_R1.fastq.sorted.hc.gVCF.raw_variants.vcf
The difference is probably due to bwa and GATK behaviour. GATK we chunked into 20 pieces, while we used 1000 before. The reason GATK on Arvados is so fast is because it is using a distributed file system. Adding GATK chunks would probably bring processing time further down.
The next exercise is to 8 samples in parallel and see if that performs in a flat way.
The Arvados people will be in Leiden 9-11 June. I am going to be there too (part of the time).
11 First days in Memphis TN
Finally made it to Memphis to meet with the team and set priorities. Basically we aim to get GN2 live as soon as possible which implies a feature freeze and sharing the database with GN1 (so some functionality can be shared).
12 Installing CUDA on Penguin
The Penguin server has a Tesla K20Xm. I thought it would be a great idea to try using that GPU from Python. We'll use CUDA since it is the implementation by NVIDIA. OpenCL is catching up, so may want to check that out later (I like FOSS better).
12.1 DEFERRED Try OpenCL libraries
It also makes sense to use CUDA from a Docker container. One solution is described here.
12.2 TODO Install CUDA environment in Docker
First step is to install the CUDA driver and software provided by NVIDIA.
Fetch the driver (71 Mb) and select the right GPU and Linux. This part one needs to agree to the NVIDIA software license agreement. This means we can not distribute the driver (and probably not the toolkit).
The toolkit is a 1Gb download and binary install. By default it installs in /usr/local/cuda-7.0 which is OK on Penguin. Luckily this download link could be used from the server directly.
Installing the CUDA driver it complained there was another version 319.32. I had that overwritten with version 346.59. Also the Noveau driver needed to be removed. I don't suppose we use Penguin as a desktop, so I did that too by hand:
rmmod nouveau
The CUDA driver installer also overwrote some settings in /etc adding a file modprobe.d/nvidia-installer-disable-nouveau.conf which blacklists the driver. The kernel log says
[15874748.037936] nvidia: module license 'NVIDIA' taints kernel. [15874748.037941] Disabling lock debugging due to kernel taint [15874748.040967] nvidia: module verification failed: signature and/or required key missing - tainting kernel [15874748.063962] [drm] Initialized nvidia-drm 0.0.0 20150116 for 0000:84:00.0 on minor 1 [15874748.063977] NVRM: loading NVIDIA UNIX x86_64 Kernel Module 346.59 Tue Mar 31 14:10:31 PDT 2015 [15874748.067999] nvidia_uvm: Loaded the UVM driver, major device number 247 [15874748.069771] nvidia_uvm: Unregistered the UVM driver
The UVM is used to share memory between the CPU and GPU in CUDA programs. We won't need that for the time being.
To install the software I used screen (the installer is probably not atomic).
Do you accept the previously read EULA? (accept/decline/quit): accept Install NVIDIA Accelerated Graphics Driver for Linux-x86_64 346.46? ((y)es/(n)o/(q)uit): no Do you want to install the OpenGL libraries? ((y)es/(n)o/(q)uit) [ default is yes ]: Install the CUDA 7.0 Toolkit? ((y)es/(n)o/(q)uit): y Enter Toolkit Location [ default is /usr/local/cuda-7.0 ] Do you want to install a symbolic link at /usr/local/cuda? ((y)es/(n)o/(q)uit): y
Driver: Not Selected Toolkit: Installed in /usr/local/cuda-7.0 Samples: Installed in /home/pjotr, but missing recommended libraries Please make sure that - PATH includes /usr/local/cuda-7.0/bin - LD_LIBRARY_PATH includes /usr/local/cuda-7.0/lib64, or, add /usr/local/cuda-7.0/lib64 to /etc/ld.so.conf and run ldconfig as root Logfile is /tmp/cuda_install_31036.log
I copied the log to my Download folder and
/usr/local/cuda-7.0/bin/nvcc --version nvcc: NVIDIA (R) Cuda compiler driver Copyright (c) 2005-2015 NVIDIA Corporation Built on Mon_Feb_16_22:59:02_CST_2015 Cuda compilation tools, release 7.0, V7.0.27
Next we try to get pycuda up and running. am using the GNU Guix Python, so
guix package -i python2-virtualenv mkdir ~/guix-python virtualenv ~/guix-python
Now
export CUDA_ROOT=/usr/local/cuda-7.0 export PATH=$CUDA_ROOT/bin:$PATH ./guix_python/bin/pip install numpy # or guix install ./guix_python/bin/pip install pycuda
The install did warn that a default install may fail. Running the following example crashed the machine. Great.
export PYTHON_PATH=/home/pjotr/./guix_python/lib/python2.7/site-packages:$HOME/.guix-profile/lib/python2.7 ~/guix_python/bin/python ~/Download/pycuda-2014.1/examples/hello_gpu.py
13 Arvados
This week I was caught up in an Arvados pipeline deployment. More on that later.
14 The webserver environment
Currently GN2 has quite a few hard coded paths in the repository. Most of these paths are local to the git repository - exceptions being references to outside modules for gemma, plink, and R/qtl. These should be captured in a config file.
GN2 already has a settings file which is read in at startup and another default in the git repo in wqflasq/./cfg/defaultsettings.py. In the webserver these settings are picked up with app.config['LOGFILE']. Adding settings to the config they simply show up. It makes sense to have a PYLMMPATH setting, e.g.
PYLMM_PATH = '/home/(...)/pylmm'
To locate the module. That way it can be placed anywhere.
Which I now pick up with something like
if os.environ['PYLMM_PATH'] is None:
PYLMM_PATH=app.config['PYLMM_PATH']
if PYLMM_PATH is None:
PYLMM_PATH=os.environ['HOME']+'/gene/wqflask/wqflask/my_pylmm/pyLMM'
if !os.path.isfile(PYLMM_PATH+'/lmm.py'):
raise 'PYLMM_PATH unknown or faulty'
PYLMM_COMMAND= 'python '+PYLMM_PATH+'/lmm.
At runtime the loaded module path can also be found and set with
cwd = os.path.dirname(unicode(__file__, encoding)) sys.path.insert(1,cwd)
I also started on creating a reproducible Guix environment for pylmm. The first are scipy and numpy which install fine.
guix package -i python-2.7.6 python2-scipy python2-numpy python-simplejson
This should be enough to run pylmm, but for the GN2 interface simplejson and redis modules are required. So running
env PYTHONPATH=/home/pjotr/.guix-profile/lib/python2.7/site-packages ~/.guix-profile/bin/python $pylmm_lib_path/runlmm.py --pheno data/small.pheno --geno data/small.geno run
gives
ImportError: No module named redis
Almost there for reproducible pylmm. Especially since Redis is not required outside GN2.
15 Fixing the error
Good news, the latest pylmm/GN2 runs in GN2.
The TypeError bug was fixed. It was due to a data conversion from the webserver, something that I could not see because it was not replicated in my regression tests and because these tests don't use the webserver. Since yesterday I am running GN2, so from now on I can try on the webserver first before pushing to staging - this should speed integration up.
Meanwhile, Artem created scripts for a Docker image for GN2.
16 Replicating the error
Artem and I managed to install and run GN2 (he locally with a small test database and me on Penguin). The installation process needs work for future deployment in particular where it comes to local settings.
16.1 TESTING Improve deployment of GN2
The first error is
File "/home/pjotr/izip/git/opensource/python/gn2/wqflask/wqflask/my_pylmm/pyLMM/lmm.py", line 65, in <module> import standalone as handlers ImportError: No module named standalone
Oh great, that is mine. So I need to add that to the Python path.
(ve27).../gn2/wqflask$ env PYTHONPATH=$PYTHONPATH:.../gn2/wqflask/wqflask/my_pylmm/pyLMM python ./runserver.py
That works, but now it turns out several things are hard coded in, for example, runserver.py. Not so great, but at least I can test and look for that bug. The regression tests all pass, even when using Zachary's Python installation. The server is running on localhost which was opened for Danny.
And, finally, I can replicate the error!
File "/home/zas1024/gene/wqflask/wqflask/my_pylmm/pyLMM/genotype.py", line 41, in normalize x = True - np.isnan(ind_g) # Matrix of True/False TypeError: Not implemented for this type
Interestingly the server never realizes there is an error, so that can be improved.
16.2 TODO Make sure the server immediately realizes an error
17 Fixing bugs
Next step is to replicate the pylmm/GN2 type error which strangely shows with the webserver setup and not with mine. I guess I have to copy Zachary's environment to duplicate the setup.
18 Changing environments
I am creating my own setup (on my laptop and Penguin) for development. GNU Guix is running now which means I have the latest development software at my finger tips. In the installation process I worked out an idea for a 1-step install of GNU Guix that was followed up in the GNU Guix project. See this thread and the result. This will come in handy with CUDA installations (e.g. on Ubuntu we need a different gcc compiler). For example, on Penguin the default apt-get wants to install a whole range of new packages including
consolekit cpp-4.4 dkms g++-4.4 gcc-4.4 gcc-4.4-base lib32gcc1 libboost-python1.46.1 libboost-thread1.46.1 libc-bin libc-dev-bin libc6 libc6-dev libc6-i386 libck-connector0 libcublas4 libcudart4 libcufft4 libcurand4 libcusparse4 libdrm-dev libdrm-nouveau2 libgl1-mesa-dev libjs-sphinxdoc libnpp4 libpam-ck-connector libpolkit-agent-1-0 libpolkit-backend-1-0 libqtassistantclient4 libstdc++6-4.4-dev libthrust-dev libvdpau-dev libvdpau1 libxext-dev libxkbfile1 libxvmc1 mesa-common-dev nvidia-304 nvidia-compute-profiler nvidia-cuda-dev nvidia-cuda-doc nvidia-cuda-gdb nvidia-cuda-toolkit nvidia-current nvidia-opencl-dev nvidia-settings opencl-headers policykit-1 policykit-1-gnome python-glade2 python-gtk2 python-mako python-markupsafe python-matplotlib python-pycuda-doc python-pycuda-headers python-pytools python-xkit screen-resolution-extra x11-xkb-utils x11proto-xext-dev xfonts-base xserver-common xserver-xorg-core
This is not funny on a running server!
GNU Guix resolves this by clean separation of software packages. Guix, at this point, has no CUDA support, but Nix has a CUDA package, so it should be doable.
18.1 TODO Create python-CUDA package for GNU Guix
19 Running pylmm/GN2 on Penguin
Now with access to 32-core Penguin it is interesting to run current pylmm/GN2 again:
Command being timed: "env PYTHONPATH=my_pylmm/pyLMM:./lib python my_pylmm/pyLMM/runlmm.py --pheno test.pheno --geno test.geno run" User time (seconds): 588.99 System time (seconds): 79.33 Percent of CPU this job got: 841% Elapsed (wall clock) time (h:mm:ss or m:ss): 1:19.46 Maximum resident set size (kbytes): 19408080
Effectively using 8 cores with individuals = 1219 and SNPs = 98740. This is running without Redis because Redis on Penguin complains about the object size! Strange it does not show in GN2 - maybe the database has smaller datasets.
19.1 TODO Fix Redis not accepting large objects on Penguin
20 Installing Guix on GN2/Penguin
The current goal is to fix the TypeError bug that is hampering pylmm/GN2 0.50-gn2-pre1. For this I decided to install GN2 for the first time. Danny sent some useful instructions and Lei has given me access on Penguin. I am adding an INSTALL document to the GN2 git repo.
20.1 DONE Fix pylmm/GN2 TypeError from the GN2 webserver
The first thing I do is install GNU Guix (mostly so I can have my reproducible developer environment) which is the great software package manager by the GNU project. GNU Guix does not interfere with other software installs because everything is installed in one directory (/gnu). There is a bit of excitement too because I discover Ricardo Wurmus is adding bioinformatics packages to GNU Guix, as well as R and Julia! GNU Guix is going to be very important for software deployment and reprodicibility and I hope to get the GN2 environment sorted in Guix at some point.
BTW, also added an addition to bio-vcf - one of the better tools I have written and used at the Hubrecht Institute for filtering WGS variant calls. There is also a request to add to bio-locus which is a tabix replacement and another interesting (but less mature) tool.
21 Avoiding 3x extraneous genotype matrix transpose
Transposing matrices: the current version gets the genotype matrix by individual (rows) which means the matrix is transposed thrice
- before entering pylmm/GN2 proper
- before entering the kinship calculation
- before entering the GWAS calculation
These transposes can be avoided if the data is already transposed (i.e., ordered by genotype/SNP) in the original source. In fact, it often is!
For smaller dataset it does not matter, but when we get to large data it makes sense to save on these extraneous calculations mainly because a transpose needs all genotype data in RAM. Nick's later pylmm avoids transposes by using an interator, so we are going to do it in a similar way. In the first `sprint' I made sure the extraneous transposed matrices are out of the way.
22 Optimizing pylmm/GN2
The current version of GN2 loads all the data in one Redis BLOB. To start calculations while loading the data (and limit RAM use) it would be useful to (re-introduce) iterating by genotype (SNP). Also it would be a good opportunity to introduce named genotypes (SNPs) - so we can track deleted phenotypes. For the iteration itself we are going to pass around a function that fetches the data - Python generators come in handy there.
Cutting Redis out altogether saved 5 seconds (out of 23). We'll have to see how that pans out with larger datasets. I am putting in the iterator infrastructure first.
23 pylmm home stretch (0.50 release)
News flash Julia is now part of GNU Guix. This means we can now package the ldc D compiler because llvm is supported. That is very interesting because GNU Guix plays well with Docker.
Since no one commented on the difference in genotype normalization handling I am going to default to the new version from now on. This has gone into release 0.50-gn2. See the release notes for GN2/pylmm.
24 Genotype normalization problem
In further work I tried to switch back to the old kinship calculation for testing purposes. Unfortunately the normalization differs, so I had to take it apart.
When starting with a genotype of
geno (4, 6) [[ 1. 1. 0. 1. 1. 1. ] [ 1. 1. 1. 1. 1. 1. ] [ 0.5 0. 0.5 0. 1. nan] [ 0. 0. 0. 0. 0. 0. ]]
The old GN2 genotype normalization reads for every SNP
not_number = np.isnan(genotype_matrix[:,counter])
marker_values = genotype_matrix[True - not_number, counter]
values_mean = marker_values.mean()
genotype_matrix[not_number,counter] = values_mean
vr = genotype_matrix[:,counter].var()
if vr == 0:
continue
keep.append(counter)
genotype_matrix[:,counter] = (genotype_matrix[:,counter] - values_mean) / np.sqrt(vr)
which results in
G (after old normalize) (6, 4) = [ [ 0.905 0.905 -0.302] ... [ 0.905 -0.302 -1.508] [ 1. 1. -1.] ... [ 1. -1. -1.] ... [ 0.577 0.577 0.577] ... [ 0.577 0.577 -1.732] [ 0.816 0.816 0. ] ... [ 0.816 0. -1.633] ]
Meanwhile the following genotype normalization code is part of the new pylmm
missing = np.isnan(g)
values = g[True - missing]
mean = values.mean() # Global mean value
stddev = np.sqrt(values.var()) # Global stddev
g[missing] = mean # Plug-in mean values for missing data
if stddev == 0:
g = g - mean # Subtract the mean
else:
g = (g - mean) / stddev # Normalize the deviation
return g
which results in
G (6, 4) = [ [ 0.905 0.905 -0.302] ... [ 0.905 -0.302 -1.508] [ 1. 1. -1.] ... [ 1. -1. -1.] ... [ 0.577 0.577 0.577] ... [ 0.577 0.577 -1.732] [ 0.707 0.707 0. ] ... [ 0.707 0. -1.414] ]
where notably the last line differs. Correct me if I am wrong, but I believe the old GN2 genotype normalization code gets it wrong.
25 Starting up again (Sambamba on Google OSS BLOG)
News flash Artem's Sambamba gets a page on Google's open source blog!
Back on the job after a week of transition (from Africa to the North of the Netherlands). Today I pushed pylmm release 0.50-gn2-pre1 to github master for testing.
26 RSS feed fix
The RSS feed should be working properly now. You may see duplicate entries for the older feed items. That will resolve.
27 Plot of timings
Today some plotting with matplotlib/py to plot performance increases of pylmm so far. Also a good moment to experiment a bit with Pandas. I have a testing repository which contains a script that will run a performance test and plot the results.
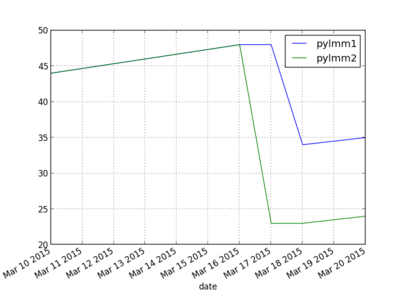
The plot (X-axis time in seconds) is interesting because the tests first got slower when I introduced multi-core Kinship support on 16/3 (pylmm1 got updated too, I'll regress that soon) - the multi-core Kinship version does not pay with this small dataset of 1K individuals by 8K SNPs. 17/3 GWAS multi-core got introduced in pylmm2. 18/3 the number of Redis updates for the progress meter to the website got reduced in pylmm1. 20/3 both got a bit slower because of an upgrade of numpy.
These results are for a MacBook Pro. Next week I'll update that for the server and hopefully we can start using a larger test set.
28 Preparing for adding Human GWAS support
So far I have worked on the `runother' function and now it was time to look at the `runhuman' edition. First difference is that covariates are passed in and genotypes as a plink filename rather than a genotype matrix. Also the function handles missing phenotype and genotype data. For the rest the only difference is calling a `humanassociation' function which takes care of the added covariates.
By the looks of it we can treat all GWAS in a unified way. First add covariates and the plink file handler.
28.1 TODO Add data human and testing framework
28.2 TODO Add covariate handler
28.3 TODO Handle plink input file
Meanwhile, Artem is playing around with GWAS.
And Zachary and Danny are integrating my LMM updates in small incrementals. There are two improvements coming for the passing of data between the webserver and pylmm. First is that we need to pass in the names of the SNPs, phenotypes and individuals. The reason being that pylmm can check whether the data matches between items and that I will return data unordered and trimmed (missing values). The only way for the webserver to know what is what is by having the matching names. The second improvement is that Redis should pass the data by SNP, rather than one genotype block. This allows iteration over the data and speed up processing (on multi-core).
Python's numpy does not properly supports coupling of named tablerows and columns. A different module could be Pandas. Fortunately there are only a few places where matrices/vectors get trimmed/reordered, so we may just inject that support and forget about making things beautiful. Beauty tends to come at a cost too…
29 Adding the progress meter
GN2 uses Redis to communicate progress to the web server. In the previous implementation the calculation code was `sprinkled' with Redis updates and print statements, such as here. This screams for a lambda solution because the calculation should not care about progress updates. Same story for debugging output. So I am going to introduce (essentially) three functions that get passed around as callbacks which should also take care of the existing stderr/stdout mix and will allow us to tweak output depending on use.
The debug output should align with Python's logging capability. Stdout output, meanwhile, is simple enough (though one may want to write to file later). The progress function gets (function) location, line, and total as parameters. I think the new usage is more pleasing to the eyes than the old usage. The implementation is here.
Interestingly, the original GN2/pylmm implementation got faster - from 44 seconds down to 35 seconds. This because now we don't update the Redis progress meter at every iteration, but only when the value changes properly. Updating Redis is expensive. The timings for the new multi-core version remain the same since I had no progress meter in there. Now that is added too.
See the release notes for GN2/pylmm 0.50-gn2-pre2.
30 Adding multi-core GWAS into GN2
The first multi-core GN2/pylm version 0.50-gn2-pre1 has been released on my lmm branch:
I have been adding the multi-core GWAS code to the lmm branch on github. The good news is that the time is halved on my dual-core from 44 seconds to 23:
[40%] Calculate Kinship: 6.65058517456 [60%] Doing GWAS: 9.86989188194 User time (seconds): 55.17 System time (seconds): 1.94 Percent of CPU this job got: 244% Elapsed (wall clock) time (h:mm:ss or m:ss): 0:23.32 Maximum resident set size (kbytes): 1170060 Minor (reclaiming a frame) page faults: 1233496 Voluntary context switches: 15309 Involuntary context switches: 3876
The results are the same as before using the later pylmm calculations. It is still possible to run both the old and the new versions of pylmm through GN2. The main differences are
- The new version is multi-core
- The new version is based on the updated version by Nick
- The new version removes missing phenotypes
Interestingly, when comparing the older GN2 and the later version of pylmm on single core, the latter is quite a bit slower:
Old (GN2/pylmm) [23%] Calculate Kinship: 6.79350018501 [77%] Doing GWAS: 22.3544418812 New pylmm [5%] Calculate Kinship: 6.60184597969 [95%] Doing GWAS: 128.968981981
Using the 2 cores (4 hyperthreaded) you get
[40%] Calculate Kinship: 6.74116706848 [60%] Doing GWAS: 9.94114780426
So, now it is twice as fast. I bet a study of the code with a profiler offers scope for speedups.
30.1 DONE Profile the new GWAS computation
And, now we can add this GWAS calculation to GN2. There are a few small TODOs before inclusion into GN2:
30.2 DONE Callback for progress meter
30.3 IN PROGRESS Make sure SNPs are named (because order may differ)
Actually, I kinda solved that problem by sorting the output - but that will be expensive with really large datasets.
Thereafter we continue with speed optimizations (and RAM!).
31 Performance stuff and adding multi-core Kinship into GN2
I have added the multi-core Kinship calculation to GN2. Rather than hack the existing code I provide a bypass. The original GN2/Redis based test (98740 snps, 1219 inds) would not run on my laptop (usurping all 8GB of RAM) within the GN2/pylmm code base. So I used the reduced (8000 snps, 1219 inds) for testing purposes instead:
[29%] Calculate Kinship: 10.0904579163 [6%] Create LMM object: 1.95971393585 [0%] LMM_ob fitting: 0.0321090221405 [65%] Doing GWAS: 22.7719459534 Command being timed: "env PYTHONPATH=my_pylmm/pyLMM:./lib python my_pylmm/pyLMM/runlmm.py --geno data/test8000.geno --pheno data/test8000.pheno redis --skip-genotype-normalization" User time (seconds): 41.89 System time (seconds): 1.10 Percent of CPU this job got: 97% Elapsed (wall clock) time (h:mm:ss or m:ss): 0:44.30 Maximum resident set size (kbytes): 1076584 Minor (reclaiming a frame) page faults: 513535 Voluntary context switches: 31917 Involuntary context switches: 787
Nick's original pylmm took 15 seconds to calculate K and 28 seconds to calculate GWAS. That (kinda) compares with the above (though the Redis slowdown is not shown here). Replacing the Kinship calculation with the multi-core one:
[20%] Calculate Kinship: 6.36837601662 [6%] Create LMM object: 2.0025908947 [0%] LMM_ob fitting: 0.0384421348572 [73%] Doing GWAS: 22.6719589233 User time (seconds): 57.58 System time (seconds): 1.86 Percent of CPU this job got: 123% Elapsed (wall clock) time (h:mm:ss or m:ss): 0:48.31 Maximum resident set size (kbytes): 1381140 Minor (reclaiming a frame) page faults: 1094998 Voluntary context switches: 25462 Involuntary context switches: 1585
is not exciting (just yet). It is faster internally (6 seconds instead of 10 seconds for K), but for this dataset slower overall. Should be much better with larger data - which I still can't run on my laptop (will fix later with:) the new code does not need a transform G matrix, so
31.1 DONE (Re-)introduce SNP-based iterator for K calculation
That requires revisiting the Redis component (also on the TODO list).
One further optimization would be to reuse the kinship matrix from GN2 which means we have to pass it back to the web application (or cache it locally - the latter is probably the best idea though I'll need a way to find out it is the same genotype matrix).
31.2 TODO Create cache for K
Artem and I have been reading on CPU optimizations and (separately) the problems around the Julia programming language. The latter is a bummer because I rather like the design of Julia. Looks like Julia is out because it is not mature enough.
Nick has just published a paper in Genetics on optimizing a scan for multiple (interacting) phenotypes with a 10-fold speed increase. Every little bit helps.
32 GN2/pylmm kinship calculation
Must be Friday the 13th. Today I am stuck in the fact that the kinship calculation in GN2 changes the genotype matrix G as a side-effect. Not only that, kinship is calculated multiple times with runother, stacking changes in G. Also it differs from the later implementation in pylmm. Doing a naive kinship calculation differs again. So, to check what it does, let me do it by hand:
Take a genotype array with 4 individuals by 5 genotypes (A=0, H=.5 and B=1):
> g
[,1] [,2] [,3] [,4]
[1,] 1 1 0.5 0
[2,] 1 1 0.0 0
[3,] 0 1 0.5 0
[4,] 1 1 0.0 0
[5,] 1 1 1.0 0
and its transpose
> t(g)
[,1] [,2] [,3] [,4] [,5]
[1,] 1.0 1 0.0 1 1
[2,] 1.0 1 1.0 1 1
[3,] 0.5 0 0.5 0 1
[4,] 0.0 0 0.0 0 0
Now take the dot product
> t(g) %*% (g)
[,1] [,2] [,3] [,4]
[1,] 4.0 4 1.5 0
[2,] 4.0 5 2.0 0
[3,] 1.5 2 1.5 0
[4,] 0.0 0 0.0 0
And divide it by the number of SNPs
> (t(m) %*% m)/5
[,1] [,2] [,3] [,4]
[1,] 0.8 0.8 0.3 0
[2,] 0.8 1.0 0.4 0
[3,] 0.3 0.4 0.3 0
[4,] 0.0 0.0 0.0 0
From this it shows that individual 1 and 2 are (possibly) closely related and 4 is furthest removed. Also the kinship matrix is symmetrical.
This is what pylmm does (or should do) to calculate kinship K. To avoid loading the full matrix in RAM, Nick does the multiplication a SNP at a time - which lends itself to multi-core processing. The version in GN2, however, gives different results:
K = kinship_full(G,options) print "first Kinship method",K.shape,"\n",K K2 = calculate_kinship(np.copy(G.T),None,options) print "GN2 Kinship method",K2.shape,"\n",K2
(where calculatekinship is the GN2 method) results in
first Kinship method (4, 4) [[ 0.8 0.8 0.3 0. ] [ 0.8 1. 0.4 0. ] [ 0.3 0.4 0.3 0. ] [ 0. 0. 0. 0. ]] GN2 Kinship method (4, 4) [[ 0.79393939 0.35757576 -0.44242424 -0.70909091] [ 0.35757576 1.08484848 -0.2969697 -1.14545455] [-0.44242424 -0.2969697 0.5030303 0.23636364] [-0.70909091 -1.14545455 0.23636364 1.61818182]]
After genotype normalization, the first kinship method also prints
first Kinship method (4, 4) [[ 0.79393939 0.35757576 -0.44242424 -0.70909091] [ 0.35757576 1.08484848 -0.2969697 -1.14545455] [-0.44242424 -0.2969697 0.5030303 0.23636364] [-0.70909091 -1.14545455 0.23636364 1.61818182]]
So, now they do agree, which is a comfort. The full K for 98740 SNPs should be
(1219, 1219) [[ 1.28794483 1.28794483 1.28794483 ..., 0.10943375 0.10943375 0.10943375] ..., [ 0.10943375 0.10943375 0.10943375 ..., 1.29971401 1.29971401 1.29971401]]
Which agrees with
castelvetrano:~/izip/git/opensource/python/test_gn_pylmm$ env PYTHONPATH=$pylmm_lib_path:./lib python $pylmm_lib_path/runlmm.py --geno test.geno kinship
Meanwhile the test8000 should be
Genotype (8000, 1219) [[-2.25726341 -2.25726341 -2.25726341 ..., 0.44301431 0.44301431 0.44301431] ..., [-1.77376957 -1.77376957 -1.77376957 ..., -1.77376957 -1.77376957 -1.77376957]] first Kinship method (1219, 1219) [[ 1.43523171 1.43523171 1.43523171 ..., 0.19215278 0.19215278 0.19215278] ..., [ 0.19215278 0.19215278 0.19215278 ..., 1.26511921 1.26511921 1.26511921]]
And I confirmed the original pylmmKinship.sh does the same thing!
This week the Kinship calculation in GN2 can go multi-core.
I am reading Lippert's thesis now. He has a great way of explaining things.
32.1 DEFERRED Move W into parallel routine
32.2 DONE Finalize multi-core K into GN2
32.3 DEFERRED Does GN2 need Kve/Kva results?
33 More FaST-LMM vs pylmm
Artem is doing some excellent forensics on pylmm. At the same time I got bitten by Python's module-wide/visible outer scope in functions. I think the design is dangerous even though it can be useful as a (kind of) closure - the way I use it in plink.py fetchGenotypes(), for example. Ruby's lambda does a better job and is more consistent. In a different case I got bitten by Python's non-const list elements, when I modified them inside a function (passing the list as a reference). The default behaviour should be (deep) const. This behaviour can be emulated (expensively) by using numpy deep copying - this I will use by default from now on.
34 GN2/pylmm pull request
The GN2/pylmm code converged to these results when running from the command line in test mode:
mm37-1-3125499 0.35025804145 0.726205761108 (...) mm37-1-187576272 0.413718416477 0.679153352196 mm37-1-187586509 0.942578019067 0.346084164432
so now I have a version with Redis that can be tested against. Loading the Redis database, btw, is pretty slow. We'll work on that later.
I have created a pull request for Zachary. This version of GN2/pylmm should just work with the web server.
35 pylmm vs FaST-LMM
Artem is continuing his research on FaST-LMM and his findings chime with mine which are that pylmm is simplistic. A full comparison of FaST-LMM and pylmm can be found here. Also I have found the pylmm code to be problematic. We really ought to replace pylmm as soon as we can.
The last period I have dealt with getting data into a better file representation - that code will be useful for the future lmm resolver. It also builds on the file definitions I started for qtlHD. These are all readable tabular files which can have binary representations (if deemed useful).
Now I can read the data properly I can finally populate Redis and emulate GN2/pylmm runs. A first trial shows that kinship and pheno match, but that the genotype matrix has been normalized, i.e., pylmmGWAS creates
('kinship', array([[ 1.28794483, 1.28794483, 1.28794483, ..., 0.10943375,
0.10943375, 0.10943375],
[ 1.28794483, 1.28794483, 1.28794483, ..., 0.10943375,
0.10943375, 0.10943375],
...,
[ 0.1277144 , 0.1277144 , 0.1277144 , ..., 0.02614655,
0.02614655, 0.02614655],
[ 0.1277144 , 0.1277144 , 0.1277144 , ..., 0.02614655,
0.02614655, 0.02614655]]))
('pheno', array([ 0.57832646, 0.19578235, -0.52130538, -1.91220117, -0.12869336
,
0.34261112, 0.66442607, -1.94655064, -0.50776009, -1.23151463]))
('geno', array([[-2.25726341, 0.32398642, -4.05398355, ..., 0.22911447,
0.22752751, 0.61761096],
[-2.25726341, 0.32398642, -4.05398355, ..., 0.22911447,
0.22752751, 0.61761096],
...,
[ 0.44301431, 0.32398642, 0.24667096, ..., 0.22911447,
0.22752751, -1.61914225],
[ 0.44301431, 0.32398642, 0.24667096, ..., 0.22911447,
0.22752751, -1.61914225]]))
while my runlmm.py creates
geno [[0.0, 1.0, 0.0, 1.0, 1.0, 1.0, 1.0, 0.0, 0.0, 0.0, 0.0, 1.0, 1.0, 1.0, 1.0 , 0.0, 0.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0 , 1.0, 1.0, 1.0, 1.0, 1.0...
suggesting I need the normalisation step to replicate results. And, indeed, after normalisation we get the same results. Note the normalization happens per SNP row.
36 PLINK file formats
Back at Kilimanjaro with Marco and Sonda, continuing to hack plink file formats. I can now fetch the allele types from a plink .bed file and, using the original pylmm code, convert them to values nan, 0.0, 0.5, 1.0 for - (missing), A (hom), H (hetero) and B (hom) respectively. The code is becoming a lot simpler and cleaner.
Normalize was part of the plink class and the code does the following (comments are mine)
def normalizeGenotype(self,G):
x = True - np.isnan(G) # find non-missing values
m = G[x].mean() # calc average
s = np.sqrt(G[x].var()) # calc stddev
G[np.isnan(G)] = m # plug average in missing values
if s == 0: # if stddev is zero
G = G - m # subtract the average for all
else: # else
G = (G - m) / s # subtract the average for all and divide by stddev
return G
I can see we can plug in missing values. Subtracting the average can make sense to `normalize' the effect size and maybe the stddev can kick in too for effect comparison, but are we not talking about the genotype here? Not sure why we want to normalise allele frequencies in this way. Nick writes somewhere that it is OK to center the genotype. Meanwhile the CLI switch says By default the program replaces missing genotypes with the minor allele frequency. The –removeMissingGenotypes option (which actually sets options.normalizeGenotype) overrides that behavior by (supposedly) making the program remove missing individuals.
I need to check why that is wanted. As far as I can tell, such normalisation is not used by pylmm. Anyway, it should not be specific to plink.
36.1 DONE Check centering of genotype for GN2
37 convertlmm.py
This coming weekend I have meetings with elephant, giraffe and zebra set up. Before that I am working on GN2/pylmm. First step is to create a command line multiplexer runlmm.py so we remove command line parsing complexity from the main routines. Where Nicks code supports reading multiple input types, GN2 simply passes the data using Redis. So, I started on a tool `convertlmm.py' to map the input to a simpel textual tab delimited representation that (kinda) matches the Redis type (I like tab delimited files). There are three files, for kinship (K), genotype (G) and phenotype (Y). After conversion `runlmm.py' should simply be able to take either Redis types or these flat files.
37.1 DONE create and parse simple TSV Kinship format
Now pylmm's kinship file (.kin) is already a matrix of floats. Only problem is that it is space separated (don't do that) and does not give you the size of the matrix nor the names of the individuals. So I changed that to a header with TSV:
# Kinship format version 1.0
# Individuals = 1046
ind1 ind2 ind3
ind1 1.287 1.287 1.287 ...
ind2 0.6 0.6 0.6 ...
etc. etc.
This way the file format has some basic checking built in, also Python has a nice library named Pandas for parsing such files, but, for the time being, I'll stick to tab delimited parsers.
The kinship file does not need high precision - that would suggest that the method is precise! I'll just make sure that the values close to zero get represented well.
37.1.1 TODO Reduce precision of kinship matrix for easier storage
37.2 DONE create and parse simple TSV Phenotype format
The phenotype file (.pheno) should be individual by phenotype
# Phenotype format version 1.0
# Individuals = 1046
# Phenotypes = 2
# pheno1 = { "type": "double" }
# pheno2 = { "type": "integer" }
pheno1 pheno2
ind1 12.0 3
ind2 2.0 4
Probably we'll add some additional JSON metadata to the header later, describing the phenotype etc. Use JSON for headers, it is so much easier for to parse!
I looked at pylmm's input module to parse phenotypes, but the thing is so convoluted that it is worth rewriting. The new parsers in runlmm are not checking inputs (yet), but they do the job.
37.3 DONE create and parse simple TSV Genotype format
The genotype file (.geno) should be SNP allele by individual. Where the HAB encoding is - for missing, H for heterozygous, and A and B for homozygous:
# Genotype format version 1.0
# Individuals = 1046
# SNPs = 96500
# Encoding = HAB
ind1 ind2 ...
snp1 H A
snp2 A B
Interestingly the resulting compressed file (bzip2) is less than half the size of the plink bed format! Just goes to show one should be careful with 'optimized' binary formats. And our version has the scope of adding more alleles.
Different encodings may be introduced later and additinal JSON metadata may go into the header.
Plink bed stores individual by row. When writing the new file the matrix is transposed in RAM (not ideal for large data).
38 CUDA, TESLA K20, K40 and K80 and Fixing the RSS feed
Yesterday I sat down for a session with Brainiarc7 who does real GPU programming for NVidia. It was very informative, not least because I am starting to grasp how complicated this stuff is, even with the prior knowledge I have about hardware. Fortunately the high-level CUDA interfaces for BLAS etc. are pretty well sorted out and relatively easy to use, and for our purposes that may be good enough. One question I have is how we are going to pipeline matrix manipulations when using multiple CPU cores to the GPU (on a TESLA K20 or K40, or K80), but that will come later. Brainiarc7 also pointed out that normal GPUs are quite useless for our purposes which means I need to get a TESLA workhorse somehow for development.
Today we had our first group meeting discussing a host of potential papers including LMM. I am delighted Joep has announced he will join the virtual group as a Postdoc. I guess we are at full capacity now.
I finally figured out why the RSS updates of this BLOG are not working properly. Apparently the order of messages should be reversed (you'd expect they use the date stamp!). After some scripting with the RSS module the new feed can be found at rss. Only problem now is that the HTML is not in CDATA yet - but that I can ignore. The old feed will also keep going for the time being.
39 Test framework GN2/pylmm and runlmm.py entry point
39.1 DONE add runlmm.py with Redis info
39.2 DONE add runlmm.py with disk version of Redis info
The test frame work for GN2/pylmm works and the outcome of GN2/pylmm is comparable to my pylmm edition. There are only minor round-off differences, such as
< mm37-1-186861978 0.33073782352 0.740899555071 > mm37-1-186861978 0.330737823519 0.740899555071
Now the time has come for some major code surgery. The tool is sitting in a straightjacket now, and as long as tests keep passing everything should be fine. With GN2/pymlmm 9000 SNPs run in 33 seconds which compares fine with the single threaded pylmm. With the full SNP set, the GWAS pylmm module used by GN2 takes double the time of Nick's pylmm module, while the results compare fine. Not sure why this is, right now.
I decided to keep te old version in the main source tree so we can fall back on that any time for comparisons. This means we can have multiple algorithms for LMM supported by the webserver (FaST-LMM will be there too).
The entry point for GN2 has moved from lmm.py to runlmm.py with a –call switch that delegates into the different implementations (see runlmm.py for details). There are also other switches in use, notably –redis for the Redis-based data passing. The old lmm.py entry point still works, but will become OBSOLETE.
Before we merge into GN2 I need to test the human association code, just to make sure it still works. To account for different results order we are also returning the SNP ids.
39.3 IN PROGRESS Check difference of treatment human data in GN2/pylmm
39.4 IN PROGRESS Test human mapping for GN2
40 Moving again on GN2/pylmm
Today I got the GN2 version of pylmm (GN2/pylmm) running standalone. Now I need to write out the results so I can start changing stuff for real. I think the most tedious bit is behind. Back to real programming soon.
GN2 passes the genotype data around using a Redis server, that means the matrix is several times in RAM. Also there is an issue that GN2/pylmm can not iteratively process data while loading from RAM or disk because the matrix is passed in as a complete unit (rather then by row).
40.1 LATER Rethink Redis mode of passing data for lmm matrices
40.2 DONE Provide an iterative way of processing the main SNP matrix
41 A virtual group
I have been a bit busy with the course and setting up my own virtual group. The virtual group acts like a real group, with meetings, presentations and writing papers.
42 Some work at KEMRI
This week I am giving a workshop on sequencing @KEMRI. The program is
MO: Basics of sequencing and mapping TU: Basics of variant calling WE: Free TH: Basics of de-novo assembly FR: Basics of gaining statistical power from populations
The second day more people showed up than the first day, which is a good sign (I suppose). I also wrote a FaST-LMM grant pre-proposal and a large BLOG about the Google Summer of Code Sambamba project which led to its publication in Bioinformatics. Hopefully they'll put that up soon.
Ethan Willis from the bioinformatics program in Memphis submitted a SQL security patch to GN.
For the GN2 project it is currently hard to find the hours to get down to real coding.
Artem found that Pypy has acquired some support for GPU recently, so that may be worth testing in the near future and see how that compares to the CPython version.
42.1 DEFERRED Test pypy GPU support
43 Setting up a test framework for pylmm in GN2
Added a github repository for testing the pylmm implementation that is used in Genenetwork2. Next, started hacking the my\lmm code to support command line access again and added the multi-core version of pylmmGWAS.py to the lmm branch. The good news is that on the first run the older LMM class gives exactly the same result with the new pylmmGWAS.py. Some comfort there and it means I can start replacing parts. The second much larger run did show a difference, where 796 our of 98741 results differed. 740 of these are missing in the GN2/LMM branch. Not sure what the cause is now (looks like they are removed because of missing data), but at least one of both trees changed something.
Next step is to modify pylmmGWAS to call into the run\other method using the mouse test data so we can strictly replicate GN2's behaviour before surgical procedures start.
44 Parallel programming is hard
A bug was reported for Ruby bio-vcf where the output generator running on separate threads was overtaken by the input processor on a fast machine! Something that was not anticipated. So, a lock had to be introduced for output.
NEWS FLASH: The sambamba paper on parallel programming in D is published online!
45 Merging pylmm updates into GN2
Pylmm in GN2 has diverged quite a bit from the original pylmm. I thought I could diff a bit and merge changes, but Python does not really make it easy because of indentation between commits :(. The only viable route is merging Nicks changes into the GN2 code base - one by one. Of course, I have also changed quite a bit of code, so those changes need to be merged in by hand too. Bummer, that is more work than I thought it would be.
Best to set up a testing frame work first, before I start introducing these patches. I am creating a testing git repo outside GN for that purpose. It can be moved into GN later, if required.
46 Passing parameters with Redis
installed redis which is an easy to use key-value server integrated in GN. From Python use the redis module.
In GN2 the marker\regression module calls lmm.py with only a Redis key as an argument with a speces identifier (human or other). The key itself, generated as
key = "pylmm:input:" + temp_uuid
and points to a JSON representation of the arguments passed in by the webserver, including the phenotype vector, genotype matrix, a time stamp and a few configuration settings. All I need to figure out is how the passed in entries map against the file formats. The results are also returned in Redis using a generated key
key = "pylmm:results:" + temp_uuid
Anyway, the short of it is that I can run lmm.py independently of the webserver quite easily (which is great for testing). Also running pylmm as a separate process takes away the nightmare of running multiple threads inside a running Python webserver.
47 Google Summer of Code (GSoC)
I also added a number of project proposals for the Google Summer of Code to SciRuby and the Open Bioinformatics Foundation:
- GNU Guix packages for bioinformatics
- Local realignment support for Sambamba
- Add LMM support to SciRuby
- Fast LLVM Ruby bindings to the Julia programming language
If you have ideas we can add more. If any of these projects gets funded we may get one or more great students involved.
48 Figuring out the logic of pylmm in genenetwork
According to Zachary pylmm gets invoked from the commandline. That happens in the wqflask subdir, probably using a REST style Flask with heatmap.py and marker\regression.py. It differs from the pylmm version because is has been adapted to accept Redis input and output. I'll adapt the GN2 edition to accept both files and Redis input and output.
48.1 DONE Merge fixes by Nick into GN2
48.2 IN PROGRESS Adapt pylmm in GN2 to accept input files
48.3 DONE Make pylmm in GN2 multi-core
49 VPN and access to a CUDA machine
Today I got access to a CUDA machine (aptly named Penguin) in Memphis, so I can try some things out shortly.
In the coming days I hope to get the multi-core system sorted for genenetworks.org. CUDA comes (right) after.
50 More GPU and also Pacbio long reads for SV
This GPU BLOG is great. Djeb goes into great detail about using the GPU, and he shows the ins and outs of dealing with GPU hardware.
Another interesting link is an Illumina/Pacbio comparison for the Malaria genome.
Artem started is own blog on the math of FaST-LMM.
51 pyCUDA
I am at KEMRI/Kenya for a few weeks. There are possibilities here for working on GWAS studies in malaria and other human pathogens.
Today I looked at pyCUDA and when I get access to an NVIDIA machine I may be able to try some first pylmm GPU matrix multiplication. Tests of the multi-core pylmm version prove we can scale up to 8 cores easily before IO runs out. Larger datasets should do even better. More on that next week because it turns out GN2 uses a different command line setup than I was testing.
52 FaST-LMM the basics
Artem discovered Lippert's thesis on FaST-LMM online. It may help disentangle some of the algorithms down the line. We will use it to study the C and Python implementations.
Next is to read up on the Python code base of FaST-LMMpy. The focus will be on discovering the functionality in the C and Python implementations and trying out the test data that comes with pylmm to see the differences.
53 Pypy and pylmm first
Since pylmm is already in use I thought we could attempt to make it as fast as possible using pypy and good performance testing (within a time frame of weeks rather than months). Unfortunately, pypy is not up to it:
Installing pypy and numpy (both latest git checkouts) was a breeze. Running
/usr/bin/time -v env PYTHONPATH=. python scripts/pylmmKinship.py -v --bfile data/test_snps.132k.clean.noX out.kin --test /opt/pypy-2.5.0-linux64/site-packages/numpy/linalg/_umath_linalg.py:79: UserWarning: npy_clear_floatstatus, npy_set_floatstatus_invalid not found (...) ImportError: No module named scipy
No surprise there. A few quick tests show that pylmmGWAS does not need scipy.linalg, but it does need the scipy.stat.t functions (part of tstat called by LMM.association). Scipy is not yet supported by pypy/numpy, so it looks like most of the work is creating an alternative for scipy.stat.t. Also matrix eigh, inv, det functions will need alternative implementations or bindings.
The pylmmKinship calculation does not use these functions, so I could run it with pypy after commenting them out. The result is disheartening. Running Pypy 2.7:
User time (seconds): 431.83 System time (seconds): 6.35 Percent of CPU this job got: 290% Elapsed (wall clock) time (h:mm:ss or m:ss): 2:30.95 Maximum resident set size (kbytes): 285736
while CPython 2.7 was almost twice as fast:
User time (seconds): 221.28 System time (seconds): 3.45 Percent of CPU this job got: 229% Elapsed (wall clock) time (h:mm:ss or m:ss): 1:37.88 Maximum resident set size (kbytes): 168812
and uses far less resources! Note that the Cpython system time (IO) is half that of pypy, so there is little to be gained there. The results differ too. They are floating point rounding-off differences, a diff shows:
< 2.846997331409238940e-01 < 1.776443685985833743e-01 < 2.821442244516297326e-01 > 2.846997331409238385e-01 > 1.776443685985833187e-01 > 2.821442244516296771e-01
That is not so serious. But the rest is.
Running the profiler on the pypy and python:
Pypy 2.7:
1 0.074 0.074 25.78 25.78 pylmmKinship.py:31(<module>)
8 0 0 15.203 1.9 pylmmKinship.py:125(compute_matrixMult)
8 0 0 14.824 1.853 optmatrix.py:28(matrixMult)
8 0 0 14.824 1.853 optmatrix.py:52(matrixMultT)
8 14.824 1.853 14.824 1.853 {_numpypy.multiarray.dot}
8 0.276 0.035 7.087 0.886 pylmmKinship.py:107(compute_W)
8000 0.836 0 6.635 0.001 input.py:138(next)
8000 0.985 0 5.628 0.001 input.py:183(formatBinaryGenotypes)
64040 3.437 0 3.437 0 {_numpypy.multiarray.array}
19 0.021 0.001 2.674 0.141 __init__.py:1(<module>)
1 2.084 2.084 2.121 2.121 npyio.py:895(savetxt)
CPython 2.7:
1 0.052 0.052 15.853 15.853 pylmmKinship.py:31(<module>)
8 0 0 7.636 0.954 pylmmKinship.py:125(compute_matrixMult)
8 0 0 7.635 0.954 optmatrix.py:28(matrixMult)
8 0 0 7.635 0.954 optmatrix.py:52(matrixMultT)
8 7.635 0.954 7.635 0.954 {numpy.core._dotblas.dot}
8 0.198 0.025 6.4 0.8 pylmmKinship.py:107(compute_W)
8000 0.603 0 6.013 0.001 input.py:138(next)
8000 2.66 0 4.991 0.001 input.py:183(formatBinaryGenotypes)
1 1.271 1.271 1.308 1.308 npyio.py:844(savetxt)
It can be seen in the 4th columen that most time is lost in the matrix multiplication (which is numpy.dot). compute W meanwile is pure python and is the same speed. With the IO CPython is faster.
Pypy had ample time to 'warm up' and CPU use shows it was maxing out. The difference is that numpy.dot on CPython calls the underlying BLAS library, while pypy is running a native implementation. Running Pypy 3 is not going make a difference.
CONCLUSION: pypy at this stage won't help speed things up. With pylmmGWAS the situation is worse since we'll need to find alternative implementation of highly optimized matrix calculations. When we have these (as GPU functions), or when pypy starts supporting scipy natively, we can have another attempt.
53.1 DONE speeding up pylmm using pypy (is not possible)
54 Pypy thoughts and running the profiler
We could try and port pylmm to pypy.
Numpy support is pretty decent now. Support for linalg is missing, but I think we can do without.
It could be another quick win. pypy appears to be quite a bit faster, though the numpy libraries are already written in C, and I don't expect a 100x speedup. But 4x seems possible. Also there is a gotcha with parallel running using multiprocessing (as we use now). But I think we can tune that by running large enough jobs.
And here perhaps a good reason to skip a pypy port and move to a different language instead.
Porting pylmm to pypy may take me anything from a few days to a few weeks. Writing a new implementation in a different language will take quite a bit longer but should be our aim anyway.
Today I also ran the Python ` -m cProfile' Cprofiler over pylmm (I know, I should have done that first). For the Kinship module it quickly became clear that the W calculation should happen in threads too (this is not notable on my dual core laptop). Note that it would mean an increase in RAM use.
For pylmmGWAS the good news is that the time consuming functionality is all in the threads:
1 0.049 0.049 28.983 28.983 pylmmGWAS.py:30(<module>)
9 0.298 0.033 18.993 2.11 pylmmGWAS.py:270(compute_snp)
8999 0.274 0 18.234 0.002 lmm.py:328(association)
45502 0.04 0 14.191 0 optmatrix.py:28(matrixMult)
54602 14.178 0 14.178 0 {numpy.core._dotblas.dot}
9000 0.668 0 6.629 0.001 input.py:138(next)
9000 2.924 0 5.494 0.001 input.py:183(formatBinaryGenotypes)
9100 0.633 0 2.496 0 lmm.py:243(LL)
1 0 0 1.99 1.99 lmm.py:159(__init__)
There may be some minor speed gain possible in the LMM.association function as about 20% of time is spent outside the matrix multiplication (which we can't improve). Also some 20% of time is spent on parsing the input files which is where pypy may show some gains, especially with multiple-cores when file loading is the bottleneck.
54.1 DEFERRED pylmmKinship would benefit from moving compute W into threads too
55 Final speedups for pylmmGWAS and pylmmKinship!
- Added library detection to pylmm to see what libraries are loaded when running the program.
- Introduced an abstraction layer to handle the matrix operations
- Gained a Python speedup of 28% on pylmmKinship (that is on single core). The multi-core version completes at 1:35 minutes using 220% CPU, down from 3:34 minutes.
- Replacing BLAS with np.dot in pylmmGWAS gains little processing time on single core (simple matrices), note that this is without ATLAS or OpenBLAS which have built-in multi-threading support, so we need to test for that too in the near future. Which begs the question of what is installed and running on the GeneNetworks server?
55.1 DEFERRED Check for ATLAS/OpenBLAS version of linalg
- By removing the blocking map function, now the multi-core version of pylmmGWAS stands at 1:50 minutes at 355% CPU, over the original 3:40 minutes. That is double speed on a dual core. Perfect.
55.2 DONE Check Phenotype order does not matter in result
I am going to let pylmm lie from here on. Time to move on to FaST-LMMpy.
55.3 DONE Add pylmmKinship.py multi-core support
55.4 DONE Add pylmmGWAS.py multi-core support
56 GWAS and more on kinship
Spent some time with Francesco looking into GWAS options for his 1000 bovine population. We are thinking along the lines of creating a haplotype map based on known pedigree, complete SNP genotyping and gene expression arrays (pheno2geno).
It made me think a bit more about the kinship matrix and it appears to me that we need something more sophisticated than what pylmm offers.
Amelie suggests that using a different kinship at every position (all SNPs - excluding those in the region being tested) is considered an idea to gain power (with a risk of overfitting). She tried something similar with 30 BxD lines but it would not give reliable estimates for the variance components. Obviously this is a rich area of R&D and when there are interesting existing algorithms out there I may give it a shot. We could also look at pheno2geno to help provide a map - with a large enough population that may help. Interesting papers I ran into are Bauman:2008 `Mixed Effects Models for Quantitative Trait Loci Mapping With Inbred Strains' and Speed:2015 `MultiBLUP: improved SNP-based prediction for complex traits'.
56.1 DEFERRED Allow for multiple kinship regions within individuals
57 Looking at missing data and the Kinship calculation got faster
I realised belatedly that my regression tests contained no missing data. The code should work, but I better get it tested too! Checking out the kinship calculation in pylmm, I can see no dealing with missing values. Not sure how that pans out in the GWAS calculation later.
57.1 DONE Check what happens to missing values in kinship calculation
Another interesting tidbit is that I removed the exception test for fblas in the Kinship calculation and that shaved off 10% of processing time. Turns out that numpy.dot is faster on my laptop than fblas! I found an explanation. Numpy uses the same amount of RAM. The results are the same, so better make numpy the default then. It could well be that numpy uses fblas anyway, but there is time saved in matrix conversions. Will find out later.
Added the –blas switch to Kinship to support the older behaviour.
In pylmm, it turns out, missing values are ignored.
57.2 DONE Check what libraries numpy is using for matrix
57.3 DONE Test numpy support in the GWAS module
57.4 DEFERRED Replace more linalg functionality in the lmm with numpy
Back to missing values, sticking one into the pheno type file made a difference to the outcome. To create a new regression test I had to go back to the original code (git branches are so helpful!). The new tests compare fine with the original and parallel code, so missing phenotype values are handled in the same way.
In the process I found out that a chunk of code in the main GWAS module is never reached. As there is little git history in pylmm's repository (and few tests), there is no way to find out how this came to be.
58 Added parallel processing for the GWAS script
Today I made the GWAS module support multi-core calculation, almost 1.5x speed on the Macbook PRO (Linux) from 3:40 down to 2:34 minutes.
That implies it should scale well on multi-core machines. I am using Pythons stdlib functions and, interestingly, the thread-pool is really shared, i.e., the interpreter is only forked once per thread-pool. That means tasks can be pretty small! The downside (to keep in mind) is that within the running interpreter global and module data is not safe.
I'll have an attempt at speeding it more by reducing the number of tasks. Also the map function blocks for results, that may explain why we are not closer to 2x speed.
58.1 DONE Improve pylmmGWAS.py multi-core support
58.2 DONE Validate pylmmGWAS missing values are working properly
Furthermore:
- Added –nthreads option to pylmm so we can specify the number of cores to use.
- Looked into matplotlib which has 2600 pages of documentation(!?) to check out plotting from python as we'll need that soon enough. I used matplotlib a few years back and liked it (we worked on Ruby alternatives in interactive D3, but so far that requires quite a bit of hard tweaking - I may use that again for interactivity). Matplotlib does EPS, SVG and D3 output, with various levels of success.
- Proposed to introduce a github org account for genenetwork and friends which also allows for private repositories.
59 First Python code for parallel processing
Today I wrote some code to speed up the kinship calculation in pylmm. Using Python's 2.7 built-in multiprocessing module the pylmmKinship calculation has gone multi-core. Where before the command
python scripts/pylmmKinship.py -v --bfile data/snps.132k.clean.noX out.kin
Took 3:36 min it now takes 1:44 min on my dual-core Macbook PRO (running Linux). That is a perfect doubling of speed. It should scale well on more CPU cores.
And a News flash: The D language, next to C bindings, now has great C++ bindings, presentation at Microsoft by Walter Bright. Here are the slides. It is interesting to see what direction D is taking. I like the integration with existing C/C++/LLVM/GNU-C++ tools, it certainly holds a promise for future GPU work.
60 Working on Sambamba paper
Little progress on FaST-LMM as per . Monday was a travel day (we are in Dar es Salaam now) and TU+WE I am working on the sambamba paper to get it accepted. Hopefully that should be completed by TH.
60.1 DONE Submit revised edition
61 Running pylmm
Ran a pylmm test on my laptop (Python 2.7.3) using the command:
castelvetrano:pylmm$ python scripts/pylmmGWAS.py -v --bfile data/snps.132k.clean.noX --kfile data/snps.132k.clean.noX.pylmm.kin --phenofile data/snps.132k.clean.noX.fake.phenos out.foo Reading SNP input... Read 1219 individuals from data/snps.132k.clean.noX.fam Reading kinship... Read the 1219 x 1219 kinship matrix in 1.066s Obtaining eigendecomposition for 1219x1219 matrix Total time: 2.110 Cleaning 432 eigen values Computing fit for null model heritability=0.000, sigma=0.986 (...) SNP 98000 Time: 4:26 min.
which produces an output file containing
SNP_ID BETA BETA_SD F_STAT P_VALUE mm37-1-3125499 0.00935975972222 0.0292676382713 0.319798940915 0.749175641213 mm37-1-3125701 -0.0370328364395 0.0289123056807 -1.28086762946 0.200484197661 mm37-1-3187481 0.0347255966132 0.0286244343907 1.21314524994 0.225309702609 (etc.)
Two runs give the exact same output. pylmm is bound to a single CPU core (99%). The timings were linear (between SNPs) and RAM use was ~60Mb.
pylmm also comes with a (simple) kinship resolver. Calculating the matrix with
python scripts/pylmmKinship.py -v --bfile data/snps.132k.clean.noX out.kin Time: 3:36 min.
is also somewhat expensive, single core (99%) and taking 84 Mb RAM. Between the runs the kinship matrix is the same, but the one in the pylmm repository differs somewhat, so either there is a machine dependent randomizer involved or it was generated in a different way.
The input BIM file is similar to a MAP format where the columns reflect chromosome, SNP name, mapping pos, bp pos (negative if unused). The FAM file looks similar to the PED format.
In the next phase I am going to try Fast-LMM and see what the performance is like. Thereafter we should try the same datasets between pylmm and Fast-LMM (it will need some conversion) and check how output and performance compares.
61.1 DONE Run pylmm
61.2 DEFERRED Run Fast-LMMpy
61.3 DONE Run same data on pylmm and Fast-LMM and compare results
62 Reading on LMM (2)
News flash: The Sambamba paper has been accepted in bioinformatics (minor revisions pending). I'll have fix some of the metrics next week, running the Picard tool.
More FaST-LMM reading today:
The R/lme4 theory document goes into some detail on LMMs. CHOLMOD Sparse direct solvers are critical to performance, and interestingly there exists an NVIDIA library. pylmm does not use sparse matrices, but FaST-LMMpy does.
Next I'll try to run the pylmm and FaST-LMM software.
63 Reading on LMM (1)
I started reading FaST-LMM related papers:
- The Lippert:2011 paper suggests that beyond 10K individuals FaST-LMM is the only feasible solution. It also suggests that only a subset of SNPs is required to compute the realized relationship matrix (RRM) and estimate association p-values
- The recent Widmer et al. (2014) paper introduces PCA to estimate the genetic similarity matrix (GSM) which makes sense because using full genome SNPs to estimate the GSM will suffer from contradicting informations (some sections of the genome will be closer to another individual than others). It is interesting they negate their own paper (Lippert:2013) finding that selecting SNPs that predict the phenotype appear to be a bad subset for estimating GSM. Here, again, a subset of SNPs is suggested to compute the RRM.
In short, the use of LMMs is suggested and recent work is mostly about performance and deciding what SNPs to choose to estimate the (family) relationships.
Today I also gave a GWAS course at KCRI (Moshi, Tanzania) to the bioinformatics unit
64 Started on FaST-LMM
Today I started looking into the Microsoft FaST-LMM Python code base, which is published under an Apache2 license.
64.1 DONE read FaST-LMM Python (FaST-LMMpy) code base
``FaST-LMM, which stands for Factored Spectrally Transformed Linear Mixed Models, is a program for performing both single-SNP and SNP-set genome-wide association studies (GWAS) on extremely large data sets. This release contains the improvements described in Widmer et al. (2014), Scientific Reports 2014, and tests for epistasis (Widmar:2014).''
``FaST-LMM is an algorithm for genome-wide association studies (GWAS) that scales linearly with cohort size in both run time and memory use (Lippert:2011).''
The good news is FaST-LMMpy it appears they have already built-in some form of cluster support. That could mean we can trivially introduce multi-core computation.
64.2 DEFERRED check for FaST-LMMpy calling out to C version
An iPython description is available. I still need to step through the singlesnp code to see if and when the C version is called. The iPython doc states:
``Now you can call FaST-LMMpy with feature selection and with these PCs as covariates. Below, we show how to do this with the C++ version of FaST-LMM. Soon, this functionality will be available in the python version.''
but it is slightly out of date. Best is to set up a run and step through the code base.
64.3 DELEGATED read R lme4 code base
Another source repo and docs to read is R's lme4 package.
65 A rotating BLOG to keep track of things
Rather than updating people by E-mail I will maintain a BLOG here with short descriptions and links to code and documentation. The first task was to set up the BLOG and RSS feed
65.1 DONE set up emacs org-mode to generate a BLOG and RSS feed
65.2 DONE improve look-n-feel of the BLOG (HTML+CSS)
The bare bone version works. In time I will improve the looks of the generated pages.
I like emacs org-mode. The tooling for org-mode is nothing short of brilliant.
BLOG look-n-feel is quite usable as of .